CULVER CITY, Calif., May 5, 2025 – ImmunityBio, Inc. (NASDAQ: IBRX), a leading immunotherapy company, today announced that the Company received a Refusal to File (RTF) letter from the U.S. Food and Drug Administration (FDA) for the supplemental biologics license application (sBLA) for use of ANKTIVA plus Bacillus Calmette-Guerin (BCG) in BCG-unresponsive non-muscle invasive bladder cancer (NMIBC) for the indication of papillary disease. This RTF letter was received despite reaching unanimous guidance and encouragement at the in-person January 2025 meeting from the leadership of the Agency, including from CBER, CDER and OCE to submit this sBLA. At this meeting all key decision makers were specifically asked and unanimously confirmed that ImmunityBio should submit the sBLA as soon as possible based on the data in the single-arm trial. Relying on this unanimous guidance the Company submitted the sBLA in March 2025. The Company has already requested an urgent meeting to resolve the inconsistencies between the directives provided at the January Meeting and receipt of the RTF letter.
ANKTIVA was approved by the FDA in 2024 with BCG for the treatment of BCG unresponsive NMIBC with Papillary tumors with CIS (Cohort A). In the same clinical trial (QUILT-3.032) long term results of patients with Papillary tumors without CIS (Cohort B), was submitted as a sBLA. The RTF letter referenced herein does not impact this prior approval of CIS +/- Papillary in BCG unresponsive patients.
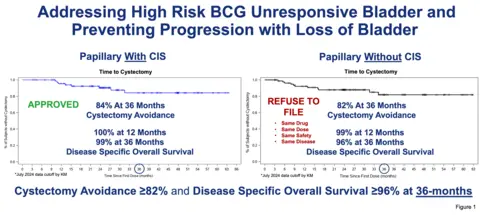
The Agency leaders present at the January meeting unambiguously invited the Company to expeditiously submit an sBLA for the NMIBC papillary indication based on the strength of the clinical response and long-term duration of follow-up data of the Papillary without CIS cohort of the QUILT 3.032 study. Data was also presented of the long term follow-up Phase 1 results which demonstrated Complete Response and Disease Free Survival in both CIS and Papillary disease, with patients in on-going cystectomy free state at 10 years. In addition disease-specific overall survival rate of 99% at 12 months and 96% at 36 months, in the BCG unresponsive patients with Papillary disease without CIS was discussed. To our knowledge, this is the most durable data to date in the Papillary setting for bladder preservation.
In March 2025, the Company completed its submission to the FDA of the sBLA for the use of ANKTIVA plus BCG in BCG-unresponsive NMIBC in the papillary indication. On May 2, 2025, and notwithstanding the FDA’s invitation to submit the sBLA articulated by its leadership at the January Meeting, the FDA delivered an RTF letter. The Company together with its consultants who attended the January 2025 meeting were shocked by this inconsistent response and have requested an urgent meeting with the Agency to resolve this issue.
“Our commitment to NMIBC patients in the papillary indication and our belief in ANKTIVA’s potential based on the strength of the clinical response and long duration of 5-year follow-up remains unchanged, despite our receipt of a refusal to file letter regarding our supplemental BLA,” said Dr. Patrick Soon-Shiong, the Company’s Founder, Executive Chairman and Global Chief Scientific and Medical Officer. “We are fully determined to work with the FDA as quickly as possible, including having already requested a Type A meeting, to explore the best path forward. We would also welcome an FDA Advisory Committee meeting as part of the regulatory process going forward. We presented our data at the recent 2025 American Urological Association (AUA) meeting and ANKTIVA+BCG was considered best in class and best in disease by the thought leaders in attendance, when compared to all the therapies currently approved or in development. Patients with BCG Unresponsive Papillary disease, face a life changing and life-threatening prospect of a total radical cystectomy, as well as the danger of the disease progressing from non-muscle invasive to muscle invasion with consequent progression and mortality. With the data presented of 99% disease specific survival at 12 months and over 82% patients not requiring bladder removal, it is essential that these patients be provided this treatment option, especially with the safety profile of ANKTIVA+BCG and the already approved indication of Papillary disease with CIS. The Company presented these data to the Agency in person in January and the Agency leaders present unanimously encouraged the company to expeditiously submit a supplemental BLA for this indication.”
Dr. Soon-Shiong further stated: “The Agency must explain the confounding inconsistency of approving ANKTIVA+BCG for patients with Papillary with CIS disease, while refusing to file the sBLA for patients with Papillary without CIS disease—even though both groups were part of the same trial, in the same high-risk population of BCG-unresponsive non-muscle invasive bladder cancer. In both cases, patients received the same surgical procedure for the Papillary component, the same therapy at the same dose, with the same excellent tolerability and meaningful clinical benefit: over 82% bladder-sparing and over 96% disease-specific survival at 36 months, as shown in Figure 1. On behalf of patients facing a potential loss of a vital organ and high risk of progression of disease, I urge the Agency to reconsider and act now.”
 “I actively participated in the January 2025 meeting at which the leadership of the Agency present at that meeting unanimously supported and encouraged ImmunityBio to submit results from the single arm trial, QUILT 3.032 as a supplemental BLA because of the high-risk of progression and metastasis these patients with BCG unresponsiveness face. The consensus was reached by all present, including me, because of the lack of satisfactory alternatives in this desperately ill population at high-risk of losing their bladder – a life threatening and life changing procedure. Thus, I was startled to learn the ImmunityBio had received a ‘Refuse to File’ letter which is in my opinion rife with regulatory inaccuracies. I recommend that the company seek a rapid meeting with the Agency to resolve this issue and minimize the delay and threat to well-being of patients who could benefit significantly from avoiding a cystectomy,” said Dr. Rachel Sherman, former FDA Principal Deputy Commissioner, who pioneered single arm trials at the height of the HIV epidemic, and was responsible for developing the expedited pathways at the Agency to address life threatening and serious diseases on behalf of the American public during her 30 year career at the agency. “Furthermore, it is incomprehensible to me that the FDA refuses to file a supplemental BLA, stating the study is not sufficient to support a regulatory review, when it has already approved a product based on that very same study in essentially the same indication and population. As stated above, these patients are suffering from a disease with a high risk of morbidity and mortality in the very short term – no delay should be tolerated,” added Dr. Sherman.
“I actively participated in the January 2025 meeting at which the leadership of the Agency present at that meeting unanimously supported and encouraged ImmunityBio to submit results from the single arm trial, QUILT 3.032 as a supplemental BLA because of the high-risk of progression and metastasis these patients with BCG unresponsiveness face. The consensus was reached by all present, including me, because of the lack of satisfactory alternatives in this desperately ill population at high-risk of losing their bladder – a life threatening and life changing procedure. Thus, I was startled to learn the ImmunityBio had received a ‘Refuse to File’ letter which is in my opinion rife with regulatory inaccuracies. I recommend that the company seek a rapid meeting with the Agency to resolve this issue and minimize the delay and threat to well-being of patients who could benefit significantly from avoiding a cystectomy,” said Dr. Rachel Sherman, former FDA Principal Deputy Commissioner, who pioneered single arm trials at the height of the HIV epidemic, and was responsible for developing the expedited pathways at the Agency to address life threatening and serious diseases on behalf of the American public during her 30 year career at the agency. “Furthermore, it is incomprehensible to me that the FDA refuses to file a supplemental BLA, stating the study is not sufficient to support a regulatory review, when it has already approved a product based on that very same study in essentially the same indication and population. As stated above, these patients are suffering from a disease with a high risk of morbidity and mortality in the very short term – no delay should be tolerated,” added Dr. Sherman.
About the QUILT-3.032 Study
QUILT-3.032 (NCT03022825) is a Phase II/III, open-label, single-arm, multicenter study of intravesical BCG plus ANKTIVA or ANKTIVA only in patients with BCG unresponsive high grade non-muscle invasive bladder cancer (NMIBC). All participants receive BCG plus ANKTIVA (Cohorts A and B) or ANKTIVA only (Cohort C) via a urinary catheter in the bladder for 6 consecutive weeks (initial induction treatment period). After the first disease assessment, eligible patients receive either a 3-week maintenance course or a 6-week re-induction course (second treatment period) at Month 3. Eligible patients will continue to receive maintenance treatment in the third treatment period at Months 6, 9, 12, and 18. Eligible patients have the option to receive maintenance treatment in the fourth treatment period at Months 24, 30, and 36. The study duration is 60 months.
Cohort A (N=100) includes patients with histologically confirmed BCG-unresponsive NMIBC CIS with or without papillary disease. The primary endpoint is biopsy-confirmed complete response (CR) rate at any time. Secondary endpoints include duration of CR, progression-free survival (PFS), time to cystectomy, safety and overall survival. FDA Approved this indication in 2024 in which eligibility included patients with Papillary disease.
Cohort B (N=80) enrolled participants with histologically confirmed BCG-unresponsive high-grade Papillary disease without CIS. The primary endpoint is a response of a disease-free rate at 12 months. Secondary endpoints include disease-free survival DFS and disease-specific survival, PFS, time to cystectomy, safety and overall survival.
About ANKTIVA®
The cytokine interleukin-15 (IL-15) plays a crucial role in the immune system by affecting the development, maintenance, and function of key immune cells—NK and CD8+ killer T cells—that are involved in killing cancer cells. By activating NK cells, ANKTIVA overcomes the tumor escape phase of clones resistant to T cells and restores memory T cell activity with resultant prolonged duration of complete response.
ANKTIVA is a first-in-class IL-15 receptor agonist IgG1 fusion complex, consisting of an IL-15 mutant (IL-15N72D) fused with an IL-15 receptor alpha, which binds with high affinity to IL-15 receptors on NK, CD4+, and CD8+ T cells. This fusion complex of ANKTIVA mimics the natural biological properties of the membrane-bound IL-15 receptor alpha, delivering IL-15 by dendritic cells and drives the activation and proliferation of NK cells with the generation of memory killer T cells that have retained immune memory against these tumor clones. The proliferation of the trifecta of these immune killing cells and the activation of trained immune memory results in immunogenic cell death, inducing a state of equilibrium with durable complete responses. ANKTIVA has improved pharmacokinetic properties, longer persistence in lymphoid tissues, and enhanced anti-tumor activity compared to native, non-complexed IL-15 in-vivo.
ANKTIVA was approved by the FDA in 2024 with BCG for the treatment of adult patients with BCG-unresponsive non-muscle invasive bladder cancer with CIS with or without papillary tumors. For more information, visit ImmunityBio.com (Founder’s Vision) and Anktiva.com.
About ImmunityBio
ImmunityBio is a vertically integrated biotechnology company developing next-generation therapies and vaccines that bolster the natural immune system to defeat cancers and infectious diseases. The Company’s range of immunotherapy and cell therapy platforms, alone and together, act to drive and sustain an immune response with the goal of creating durable and safe protection against disease. Designated an FDA Breakthrough Therapy, ANKTIVA is the first FDA-approved immunotherapy for non-muscle invasive bladder cancer CIS that activates natural killer cells, T cells, and memory T cells for a long-duration response. The Company is applying its science and platforms to treating cancers, including the development of potential cancer vaccines, as well as developing immunotherapies and cell therapies that we believe sharply reduce or eliminate the need for standard high-dose chemotherapy. These platforms and their associated product candidates are designed to be more effective, accessible, and easily administered than current standards of care in oncology and infectious diseases. For more information, visit ImmunityBio.com (Founder’s Vision) and connect with us on X (Twitter), Facebook, LinkedIn, and Instagram.
References:
https://pmc.ncbi.nlm.nih.gov/articles/PMC10813486/
Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995, such as statements regarding discussions and meetings with the U.S. FDA, the company’s submission of the sBLA for use of ANKTIVA plus BCG in BCG-unresponsive NMIBC for the indication of papillary disease and potential results therefrom, the FDA’s delivery of a refusal to file letter regarding the aforementioned sBLA submission, potential next steps, decisions and timeline related and requirements thereof, potential Type A meeting with the FDA regarding the sBLA and timing thereof and results therefrom, potential Advisory Committee meeting with the FDA regarding the sBLA and timing thereof and results therefrom, clinical trial data and potential results to be drawn therefrom, the development of therapeutics for cancer and infectious diseases, potential benefits to patients, potential treatment outcomes for patients, the described mechanism of action and results and contributions therefrom, potential future uses and applications of ANKTIVA and use in cancer vaccines and across multiple tumor types, and ImmunityBio’s approved product and investigational agents as compared to existing treatment options, among others. Statements in this press release that are not statements of historical fact are considered forward-looking statements, which are usually identified by the use of words such as “anticipates,” “believes,” “continues,” “goal,” “could,” “estimates,” “scheduled,” “expects,” “intends,” “may,” “plans,” “potential,” “predicts,” “indicate,” “projects,” “is,” “seeks,” “should,” “will,” “strategy,” and variations of such words or similar expressions. Statements of past performance, efforts, or results of our preclinical and clinical trials, about which inferences or assumptions may be made, can also be forward-looking statements and are not indicative of future performance or results. Forward-looking statements are neither forecasts, promises nor guarantees, and are based on the current beliefs of ImmunityBio’s management as well as assumptions made by and information currently available to ImmunityBio. Such information may be limited or incomplete, and ImmunityBio’s statements should not be read to indicate that it has conducted a thorough inquiry into, or review of, all potentially available relevant information. Such statements reflect the current views of ImmunityBio with respect to future events and are subject to known and unknown risks, including business, regulatory, economic and competitive risks, uncertainties, contingencies and assumptions about ImmunityBio, including, without limitation, (i) risks and uncertainties regarding commercial launch execution, success and timing, (ii) risks and uncertainties regarding market access initiatives and timing, (iii) whether the FDA will accept the Company’s request for a Type A meeting and/or Advisory Committee Meeting, (iv) whether the FDA will permit the resubmission of the sBLA and the requirements thereof, (v) uncertainties regarding the timeline of the FDA’s review of these submissions even if accepted for review and filing, (vi) whether the FDA will ultimately approve the sBLA, or other submissions in a timely matter, or at all, of which there can be no assurance, (vii) risks and uncertainties regarding limited resources at the FDA and potential delays associated therewith, (viii) whether clinical trials will result in registrational pathways and the risks and uncertainties regarding the regulatory submission, filing, review and approval process, (ix) whether clinical trial data will be accepted by regulatory agencies, (x) the ability of ImmunityBio to continue its planned preclinical and clinical development of its development programs through itself and/or its investigators, and the timing and success of any such continued preclinical and clinical development, patient enrollment and planned regulatory submissions, (xi) potential delays in product availability and regulatory approvals, (xii) ImmunityBio’s ability to retain and hire key personnel, (xiii) ImmunityBio’s ability to obtain additional financing to fund its operations and complete the development and commercialization of its various product candidates, (xiv) potential product shortages or manufacturing disruptions that may impact the availability and timing of product, (xv) ImmunityBio’s ability to successfully commercialize its approved product and product candidates, (xvi) ImmunityBio’s ability to scale its manufacturing and commercial supply operations for its approved product and future approved products, and (xvii) ImmunityBio’s ability to obtain, maintain, protect, and enforce patent protection and other proprietary rights for its product candidates and technologies. More details about these and other risks that may impact ImmunityBio’s business are described under the heading “Risk Factors” in the Company’s Form 10-K filed with the U.S. Securities and Exchange Commission (SEC) on March 3, 2025, and in subsequent filings made by ImmunityBio with the SEC, which are available on the SEC’s website at www.sec.gov. ImmunityBio cautions you not to place undue reliance on any forward-looking statements, which speak only as of the date hereof. ImmunityBio does not undertake any duty to update any forward-looking statement or other information in this press release, except to the extent required by law.
Media Contact:
Jen Hodson
Jen@nant.com


